
You are currently browsing the category archive for the ‘Biostatistics’ category.
You can install the StatRep package by downloading statrep.zip from support.sas.com/StatRepPackage, which contains:
- doc/statrepmanual.pdf – The StatRep User’s Guide (this manual)
- doc/quickstart.tex – A template and tutorial sample LATEX file
- sas/statrep_macros.sas – The StatRep SAS macros
- sas/statrep_tagset.sas – The StatRep SAS tagset for LaTeX tabular output
- statrep.ins – The LATEX package installer file
- statrep.dtx – The LATEX package itself
Unzip the file statrep.zip to a temporary directory and perform the following steps:
- Step 1: Install the StatRep SAS Macros: Copy the file statrep_macros.sas to a local directory. If you have a folder where you keep your personal set of macros, copy the file there. Otherwise, create a directory such as C:\mymacros and copy the file into that directory.
- Step 2: Install the StatRep LaTeX Package: These instructions show how to install the StatRep package in your LATEX distribution for your personal use.
- a. For MikTEX users: If you do not have a directory for your own packages, choose a directory name to contain your packages (for example, C:\localtexmf). In the following instructions, this directory is referred to as the “root directory”.
- b. Create the additional subdirectories under the above root directory: tex/latex/statrep. Your directory tree will have the following structure: root directory/tex/latex/statrep.
- c. Copy the files statrep.dtx, statrep.ins, statrepmanual.pdf, and statrepmanual.tex to the statrep subdirectory.
- d. In the command prompt, cd to the statrep directory and enter the following command: pdftex statrep.insThe command creates several files, one of which is the configuration file, statrep.cfg.
- Step 3: Tell the StatRep Package the Location of the StatRep SAS Macros. Edit the statrep.cfg file that was generated in Step 2d so that the macro \SRmacropath contains the correct location of the macro file from step 1. For example, if you copied the statrep_macros.sas file to a directory named C:\mymacros, then you de- fine macro \SRmacropath as follows: \def\SRmacropath{C:/mymacros/statrep_macros.sas} Use the forward slash as the directory name delimiter instead of the backslash, which is a special character in LaTeX.
You can now test and experiment with the package. Create a working directory, and copy the file quickstart.tex into it. To generate the quick-start document:
- Compile the document with pdfLATEX. You can use a LATEX-aware editor such as TEXworks, or use the command-line command pdflatex. This step generates the SAS program that is needed to produce the results.
- Execute the SAS program quickstart_SR.sas, which was automatically created in the preceding step. This step generates the SAS results that are requested in the quick-start document.
- Recompile the document with pdfLATEX. This step compiles the quick-start document to PDF, this time including the SAS results that were generated in the preceding step. In some cases listing outputs may not be framed properly after this step. If your listing outputs are not framed properly, repeat this step so that LaTeX can remeasure the listing outputs.
Please refer to the following file for detailed information:
Click to access statrepmanual.pdf
The first colloquium speaker at this semester, professor Wei Zheng from IUPUI, will give a talk on “Universally optimal designs for two interference models“. In this data explosive age, people are easy to get big data set, which renders people difficult to make inferences from such massive data. Since people usually think that with more data, they have more chance to get more useful information from them, lots of researchers are struggling to achieve methodological advancements under this setup. This is a very challenging research area and of course very important, which in my opinion needs the resurgence of mathematical statistics by borrowing great ideas from various mathematical fields. However, another great and classical statistical research area should come back again to help statistical inference procedures from the beginning stage of data analysis, collecting data by design of experiments so that we can control the data quality, usefulness and size. Thus it’s necessary for us to know what is optimal design of experiments. Here is an introduction to this interesting topic.
In statistics, we have to organize an experiment in order to gain some information about an object of interest. Fragments of this information can be obtained by making observations within some elementary experiments called trials. The set of all trials which can be incorporated in a prepared experiment will be denoted by 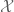

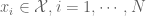
We shall restrict our attention to the parametric situation in the case of a regression model, the mean response function is then parameterized as
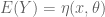
specifying for a particular 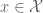

A design is specified by an initially arbitrary measure 

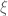
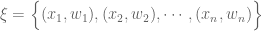
where the 

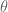

where 

Now we have to propose the statistical criteria for the optimum. It is known that the least squares estimator minimizes the variance of mean-unbiased estimators (under the conditions of the Gauss–Markov theorem). In the estimation theory for statistical models with one real parameter, the reciprocal of the variance of an (“efficient”) estimator is called the “Fisher information” for that estimator. Because of this reciprocity, minimizing the variance corresponds to maximizing the information. When the statistical model has several parameters, however, the mean of the parameter-estimator is a vector and its variance is a matrix. The inverse matrix of the variance-matrix is called the “information matrix”. Because the variance of the estimator of a parameter vector is a matrix, the problem of “minimizing the variance” is complicated. Using statistical theory, statisticians compress the information-matrix using real-valued summary statistics; being real-valued functions, these “information criteria” can be maximized. The traditional optimality-criteria are invariants of the information matrix; algebraically, the traditional optimality-criteria are functionals of the eigenvalues of the information matrix.
- A-optimality (“average” or trace)
- One criterion is A-optimality, which seeks to minimize the trace of the inverse of the information matrix. This criterion results in minimizing the average variance of the estimates of the regression coefficients.
- D-optimality (determinant)
- A popular criterion is D-optimality, which seeks to maximize the determinant of the information matrix of the design. This criterion results in maximizing the differential Shannon information content of the parameter estimates.
- E-optimality (eigenvalue)
- Another design is E-optimality, which maximizes the minimum eigenvalue of the information matrix.
- T-optimality
- This criterion maximizes the trace of the information matrix.
Other optimality-criteria are concerned with the variance of predictions:
- G-optimality
- A popular criterion is G-optimality, which seeks to minimize the maximum entry in the diagonal of the hat matrix. This has the effect of minimizing the maximum variance of the predicted values.
- I-optimality (integrated)
- A second criterion on prediction variance is I-optimality, which seeks to minimize the average prediction variance over the design space.
- V-optimality (variance)
- A third criterion on prediction variance is V-optimality, which seeks to minimize the average prediction variance over a set of m specific points.
Now back to our example, because the asymptotic covariance matrix associated with the LSE of 

and G-optimality seeks to minimize 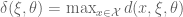
A central result in the theory of optimal design, the General Equivalence Theorem, asserts that the design 

the number of parameters.
Now the optimal design for an interference model, professor Wei Zheng will talk about, considers the following model in the block designs with neighbor effects:
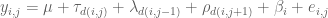
where 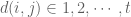

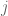

is the general mean;
is the direct effect of treatment
;
and
are respectively the left and right neighbor effects; that’s the interference effect of the treatment assigned to, respectively, the left and right neighbor plots
and
.
is the effect of the
is the random error,
.
We seed the optimal design among designs 
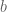
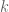
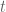
I am not going into the details of the derivation of the optimal design for the above interference model. I just sketch the outline here. First of all we can write down the information matrix for the direct treatment effect 


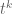

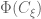
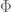
for any permutation matrix
;
is nondecreasing in the scalar
.
A measure 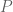
Recently some papers discussed in our journal club are focused on integrative clustering of multiple omics data sets. I found that they are all originated from factor analysis and make use of the advantage of factor analysis over principal component analysis.
Let’s recall the model for factor analysis:

where 




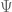
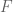


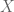

we can use the EM algorithm to get the MLE of the parameters 
Now for principal component analysis, for clarity, we are going to differentiate the classical (non-probabilistic) principal component analysis and probabilistic principal component analysis. The classical principal component analysis actually has no statistical model. And the probabilistic principal component model is defined as the above factor analysis model with 



where 




In applications, we just deal with the data matrix, 








Now think about what is the difference between the factor analysis and the probabilistic principal component analysis (PPCA). From the above definition, we see that the main difference is that factor analysis allow individual characteristics through the error term by 
with common structure 


The AOAS 2013 paper is exactly using the above idea for modeling the integrative clustering:

where 
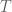

which is exactly a simple factor analysis. And this factor analysis model is more useful than PCA in this data integration setup just due to the allowing of individual characteristics for different data sources through 
The 2014 arXived paper is just generalizing the above paper by allowing another layer of individual characteristis:

But the problem for this one is how to do the estimation. Instead of using EM algorithm as used in the AOAS 2013 paper, they used the one as in the PCA by minimizing the reconstruction error.
- Interview with Nick Chamandy, statistician at Google
- You and Your Research + video
- Trustworthy Online Controlled Experiments: Five Puzzling Outcomes Explained
- A Survival Guide to Starting and Finishing a PhD
- Six Rules For Wearing Suits For Beginners
- Why I Created C++
- More advice to scientists on blogging
- Software engineering practices for graduate students
- Statistics Matter
- What statistics should do about big data: problem forward not solution backward
- How signals, geometry, and topology are influencing data science
- The Bounded Gaps Between Primes Theorem has been proved
- A non-comprehensive list of awesome things other people did this year.
- Jake VanderPlas writes about the Big Data Brain Drain from academia.
- Tomorrow’s Professor Postings
- Best Practices for Scientific Computing
- Some tips for new research-oriented grad students
- 3 Reasons Every Grad Student Should Learn WordPress
- How to Lie With Statistics (in the Age of Big Data)
- The Geometric View on Sparse Recovery
- The Mathematical Shape of Things to Come
- A Guide to Python Frameworks for Hadoop
- Statistics, geometry and computer science.
- How to Collaborate On GitHub
- Step by step to build my first R Hadoop System
- Open Sourcing a Python Project the Right Way
- Data Science MD July Recap: Python and R Meetup
- git 最近感悟
- 10 Reasons Python Rocks for Research (And a Few Reasons it Doesn’t)
- Effective Presentations – Part 2 – Preparing Conference Presentations
- Doing Statistical Research
- How to Do Statistical Research
- Learning new skills
- How to Stand Out When Applying for An Academic Job
- Maturing from student to researcher
- False discovery rate regression (cc NSA’s PRISM)
- Job Hunting Advice, Pt. 3: Networking
- Getting Started with Git
The Hardy-Weinberg equilibrium is a principle stating that the genetic variation in a population will remain constant from one generation to the next in the absence of disturbing factors. When mating is random in a large population with no disruptive circumstances, the law predicts that both genotype and allele frequencies will remain constant because they are in equilibrium.
The Hardy-Weinberg equilibrium can be disturbed by a number of forces, including mutations, natural selection, nonrandom mating, genetic drift, and gene flow. For instance, mutations disrupt the equilibrium of allele frequencies by introducing new alleles into a population. Similarly, natural selection and nonrandom mating disrupt the Hardy-Weinberg equilibrium because they result in changes in gene frequencies. This occurs because certain alleles help or harm the reproductive success of the organisms that carry them. Another factor that can upset this equilibrium is genetic drift, which occurs when allele frequencies grow higher or lower by chance and typically takes place in small populations. Gene flow, which occurs when breeding between two populations transfers new alleles into a population, can also alter the Hardy-Weinberg equilibrium.
Because all of these disruptive forces commonly occur in nature, the Hardy-Weinberg equilibrium rarely applies in reality. Therefore, the Hardy-Weinberg equilibrium describes an idealized state, and genetic variations in nature can be measured as changes from this equilibrium state.
From Hardy-Weinberg equilibrium.
PS: In a mathematical way, we have the following:

eQTL tries to regress each gene expression against each SNP, in order to find those regulatory elements. And eQTL uses “normal” samples, right? (by normal I mean “no disease” like those in 1000genome project)
GWAS compares SNPs between normal(control) and disease(test) samples, trying to find out those higher-frequency variants enriched for diseases.
linkage mapping/recombination mapping/positional cloning – rely on known markers (typically SNPs) that are close to the gene responsible for a disease or trait to segregate with that marker within a family. Works great for high-penetrance, single gene traits and diseases.
QTL mapping/interval mapping – for quantitative traits like height that are polygenic. Same as linkage mapping except the phenotype is continuous and the markers are put into a scoring scheme to measure their contribution – i.e. “marker effects” or “allelic contribution”. Big in agriculture.
GWAS/linkage disequilibrium mapping – score thousands of SNPs at once from a population of unrelated individuals. Measure association with a disease or trait with the presumption that some markers are in LD with, or actually are, causative SNPs.
So linkage mapping and QTL mapping are similar in that they rely on Mendelian inheritance to isolate loci. QTL mapping and GWAS are similar in that they typically measure association in terms of log-odds along a genetic or physical map and do not assume one gene or locus is responsible. And finally, linkage mapping and GWAS are both concerned with categorical traits and diseases.
Linkage studies are performed when you have pedigrees of related individals and a phenotype (such as breast cancer) that is present in some but not all of the family members. These individuals could be humans or animals; linkage in humans is studied using existing families, so no breeding is involved. For each locus, you tabulate cases where parents and children who do or don’t show the phenotype also have the same allele. Linkage studies are the most powerful approach when studying highly penetrant phenotypes, which means that if you have the allele you have a strong probability of exhibiting the phenotype. They can identiy rare alleles that are present in small numbers of families, usualy due to a founder mutation. Linkage is how you find an allele such as the mutations in BRCA1 associated with breast cancer.
Association studies are used when you don’t have pedigrees; here the statistical test is a logistic regression or a related test for trends. They work when the phenotype has much lower penetrance; they are in fact more powerful than linkage analysis in those cases, provided you have enough informative cases and matched controls. Association studies are how you find common, low penetrance alleles such as the variations in FGFR2 that confer small increases in breast cancer susceptibility.
In The Old Days, neither association tests nor linkage tests were “genome-wide”; there wasn’t a technically feasable or affordable way to test the whole genome at once. Studies were often performed at various levels of resolution as the locus associated with the phenotype was refined. Studies were often performed with a small number of loci chosen because of prior knowledge or hunches. Now the most common way to perform these studies in humans is to use SNP chips that measure hundreds of thousands of loci spread across the whole genome, thus the name GWAS. The reason you’re testing “the whole genome” without sequencing the whole genome of each case and control is an important point that is a separate topic; if you don’t yet know how this works, start with the concept of Linkage Disequilibrium. I haven’t encountered the term GWLS myself, but I think it’s safe to say that this is just a way to indicate that the whole genome was queried for linkage to a phenotype.
Genomic Convergence of Genome-wide Investigations for Complex Traits
###############################################################################
The following comes from Khader Shameer:
The following articles were really useful for me to understand the concepts around GWAS.
I would recommend the following reviews to understand the concept and methods. Most of these reviews refers the major studies and specific details can be obtained from individual papers. But you can get an overall idea about the concept, statistical methods and expected results from a GWAS studies from these review articles.
How to Interpret a Genome-wide Association Study
An easy to ready review article that start with basic concepts and discuss future prospects of GWAS Genome-wide association studies and beyond.
A detailed introduction to basic concepts of GWAS from the perspective of vascular disease : Genome-wide Association Studies for Atherosclerotic Vascular Disease and Its Risk Factors
Great overview of the current state of GWAS studies: Genomewide Association Studies and Assessment of the Risk of Disease
Detailed overview of statistical methods : Prioritizing GWAS Results: A Review of Statistical Methods and Recommendations for Their Application
For a bioinformatics perspective Jason Moore et.al’s review will be a good start : Bioinformatics Challenges for Genome-Wide Association Studies
Soumya Raychaudhuri’s review provides overview of various approaches for interpretations of variants from GWAS Mapping rare and common causal alleles for complex human diseases.
A tutorial on statistical methods for population association studies
Online Resources: I would recommend to start from GWAS page at Wikipedia followed by NIH FAQ on GWAS, NHGRI Catalog of GWAS, dbGAP, GWAS integrator and related question at BioStar.
##############################################################################
For introductory material, the new blog Genomes Unzipped has a couple of great posts: (From Neilfws)
I attended the The 1000 Genomes Project Community Meeting 12th and 13th July 2012 at University of Michigan. Because lots of the presentations are more from the computation point of view instead of statistical point of view, I have little idea about those talks. But from my point of view, if you want me to conclude the whole meeting into some key words, I would say “variant analysis”, “imputation”, “rare variants” and “accuracy of variant calling”.
In order to make sure I learn as most as I can from the meeting, I am going to do some post-learning work. And the note is here.
I just came back from the talk, “Statistical Methods for Analysis of Gut Microbiome Data” , given by Professor Hongzhe Lee from University of Pennysylvania.
I learned this new biological name: Microbiome—-as extended human genomes.
A microbiome is the totality of microbes, their genetic elements (genomes), and environmental interactions in a particular environment. The term “microbiome” was coined by Joshua Lederberg, who argued that microorganisms inhabiting the human body should be included as part of the human genome, because of their influence on human physiology. The human body contains over 10 times more microbial cells than human cells.
There are several research methods:
Targeted amplicon sequencing
Targeted amplicon sequencing relies on having some expectations about the composition of the community that is being studied. In target amplicon sequencing a phylogenetically informative marker is targeted for sequencing. Such a marker should be present in ideally all the expected organisms. It should also evolve in such a way that it is conserved enough that primers can target genes from a wide range of organisms while evolving quickly enough to allow for finer resolution at the taxonomic level. A common marker for human microbiome studies is the gene for bacterial 16S rRNA (i.e. “16S rDNA”, the sequence of DNA which encodes the ribosomal RNA molecule). Since ribosomes are present in all living organisms, using 16S rDNA allows for DNA to be amplified from many more organisms than if another marker were used. The 16S rDNA gene contains both slowly evolving regions and fast evolving regions; the former can be used to design broad primers while the latter allow for finer taxonomic distinction. However, species-level resolution is not typically possible using the 16S rDNA. Primer selection is an important step, as anything that cannot be targeted by the primer will not be amplified and thus will not be detected. Different sets of primers have been shown to amplify different taxonomic groups due to sequence variation.
Targeted studies of eukaryotic and viral communities are limited and subject to the challenge of excluding host DNA from amplification and the reduced eukaryotic and viral biomass in the human microbiome.
After the amplicons are sequenced, molecular phylogenetic methods are used to infer the composition of the microbial community. This is done by clustering the amplicons into operational taxonomic units (OTUs) and inferring phylogenetic relationships between the sequences. An important point is that the scale of data is extensive, and further approaches must be taken to identify patterns from the available information. Tools used to analyze the data include VAMPS, QIIME and mothur.
Metagenomic sequencing
Metagenomics is also used extensively for studying microbial communities. In metagenomic sequencing, DNA is recovered directly from environmental samples in an untargeted manner with the goal of obtaining an unbiased sample from all genes of all members of the community. Recent studies use shotgun Sanger sequencing or pyrosequencing to recover the sequences of the reads. The reads can then be assembled into contigs. To determine the phylogenetic identity of a sequence, it is compared to available full genome sequences using methods such as BLAST. One drawback of this approach is that many members of microbial communities do not have a representative sequenced genome.
Despite the fact that metagenomics is limited by the availability of reference sequences, one significant advantage of metagenomics over targeted amplicon sequencing is that metagenomics data can elucidate the functional potential of the community DNA. Targeted gene surveys cannot do this as they only reveal the phylogenetic relationship between the same gene from different organisms. Functional analysis is done by comparing the recovered sequences to databases of metagenomic annotations such as KEGG. The metabolic pathways that these genes are involved in can then be predicted with tools such as MG-RAST, CAMERA[42] and IMG/M.
RNA and protein-based approaches
Metatranscriptomics studies have been performed to study the gene expression of microbial communities through methods such as the pyrosequencing of extracted RNA. Structure based studies have also identified non-coding RNAs (ncRNAs) such as ribozymes from microbiota.[45]Metaproteomics is a new approach that studies the proteins expressed by microbiota, giving insight into its functional potential.
He analyzed two statistical methods based on the first technology listed above:
- Kernel-based regression to test the effect of Microbiome composition on an outcome
- Sparse Dirichlet-Multinomial regression for Taxon-level analysis
The following is the abstract of this talk:
With the development of next generation sequencing technology, researchers have now been able to study the microbiome composition using direct sequencing, whose output are taxa counts for each microbiome sample. One goal of microbiome study is to associate the microbiome composition with environmental covariates. In some cases, we may have a large number of covariates and identification of the relevant covariates and their associated bacterial taxa becomes important. In this talk, I present several statistical methods for analysis of the human microbiome data, including exploratory analysis methods such as generalized UniFrac distances and graph-constrained canonical correlations and statistical models for the count data and simplex data. In particular, I present a sparse group variable selection method for Dirichlet-multinomial regression to account for overdispersion of the counts and to impose a sparse group L1 penalty to encourage both group-level and within-group sparsity. I demonstrate the application of these methods with an on-going human gut microbiome study to investigate the association between nutrient intake and microbiome composition. Finally, I present several challenging statistical and computational problems in analysis of shotgun metagenomics data.
The meaning of the term ”Biological Replicate” unfortunately often does not get adequately addressed in many publications. “Biological Replicate” can have multiple meanings, depending upon the context of the study. A general definition could be that biological replicates are when the same type of organism is grown/treated under the same conditions. For example, if one was performing a cell-based study, then different flasks containing the same type of cell (and preferably the exact same lineage and passage number) which have been grown under the same conditions could be considered biological replicates of one another. The definition becomes a bit trickier when dealing with higher-order organisms, especially humans. This may be an entire discussion in and of itself, but in this case, it is important to note that one does not have a well-defined lineage or passage number for humans. Indeed, it is basically impossible to ensure that all of your samples for one treatment or control have been exposed to the same external factors. In this case, one must do all that is possible to accurately portray and group these organisms; thus, one should group according to such traits as gender, age, and other well-established cause-effect traits (smokers, heavy drinkers, etc.).
Also, it may be helpful to outline the contrast between biological and technical replicates. Though people have varying definitions of technical replicates, perhaps the purest form of technical replicate would be when the exact same sample (after all preparatory techniques) is analyzed multiple times. The point of such a technical replicate would be to establish the variability (experimental error) of the analysis technique (mass spectrometry, LC, etc.), thus allowing one to set confidence limits for what is significant data. This is in contrast to the reasoning behind a biological replicate, which is to establish the biological variability which exists between organisms which should be identical. Knowing the inherent variability between “identical” organisms allows one to decide whether observed differences between groups of organisms exposed to different treatments is simply random or represents a “true” biological difference induced by such treatment.
Biological Factor: Single biological parameter controlled by the investigator. For example, genotype, diet, environmental stimulus, age, etc.
Treatment or Treatment Level: An exact value for a biological factor; for example, stress, no-stress, young, old, drug-treated, placebo, etc.
Condition: A single combination of treatments; for example, strain1/stressed/time10, young/drug-treated, etc.
Sample: An entity which has a single condition and is measured experimentally; for example serum from a single mouse, a sample drawn from a pool of yeast, a sample of pancreatic beta cells pooled from 5 diabetic animals, the third blood sample taken from a participant in a drug study.
Biological Measurement: A value measured on a collection of samples; for example, abundance of protein x, abundance of phospho-protein y, abundance of transcript z.
Experiment: A collection of biological measurements on two or more samples.
Replicate: Two sets of measurements, either within a single experiment or in two different experiments, where measurements are made on samples in the same condition.
Technical Replicates: Replicates that share the same sample; i.e. the measurements are repeated.
Biological Replicates: Replicates where different samples are used for both replicates
Question: Technical/Biological Replicates in RNA-Seq For Two Cell Lines
I have a question around the meaning of “biological replicate” in the context of applying RNA-seq to compare two cell lines. Apologies if this is an overly naeve question.
We have two human cell lines, one of which was derived from the other. Both have different phenotypes, and we want to use RNA-seq to explore the genetic underpinnings of the difference.
If we generate one cDNA library for each sample, and sequence each library on two lanes of an Illumina GA flowcell, I understand we will have “technical replicates”. In this scenario, we can expect very little difference between the two replicates in a sample. If we were to use something like DESeq to call differential expression, it would be inappropriate to treat our technical replicates as replicates in DESeq, since that would likely lead to a large list of DE calls that don’t reflect biological differences.
So, I’d like to know if it possible within our model to have “biological replicates” with which we can use DESeq to call biologically meaningful differential expression.
So, two questions:
(1) If we grow up two sets of cells from each of our two cell lines, generate separate cDNA libraries (4 in total), and sequence them on separate lanes, would these be considered “biological replicates” in the sense that it would be appropriate to treat them as replicates within something like DESeq. I suspect not, since the fact that both replicates in a sample derive from a single cell line within a short period of time will mean that they will be very similar anyway, almost as similar as the technical replicate scenario. Perhaps we would need entirely separate cell lines to be considered biological replicates.
(2) In general, how would others address this – does it seem a better approach to go with separate cells and separate libraries, or would this entail extra effort for effectively no benefit?
Answer:
Two “biological replicate” are two samples that should be identical (as much as you can/want control) but are biologically separated (different cells, different organisms, different populations, colonies…)
You want to check the difference between cell Line A and cell line B. Let’s start assuming they are identical. Even if they are, by random fluctuation, technical issues, intrinsic slightly different environments… you will never observe that all genes have exactly the same expression. You find differences but can’t conclude if they are inevitable fluctuation or result of an actual difference.
So, you want to have 2 independent populations from A and two independent populations from B and then see how the variability WITHIN A1 and A2 compare to B1 and B2. The RNA levels from A1 and A2 WILL NOT be the same because… because biological system are far from being deterministic. They might be very similar, but different.
because A and B would be on different plates (their environment) I would seed A1 and A2, B1 and B2 the same day on 4 distinct (but as similar as possible) dishes, grow them together in the same condition to minimize external influence, and then collect at once from the 4 cell lines, extract RNA…
Since the cost is not growing cell lines, but sequencing, I would recommend to do 4 independent replicates for A and for B (or any other cell lines you may be interested in) in ONE GO, and then freeze the sample or the RNA. Even better, if you could have somebody to give you the lines called alfa, bravo, charlie, delta… (make sure they keep track of what they are in a safe place 😉 ) so that you are not biased while seeding, growing and manipulating the lines, that would be even better!
Recently we are holding a journal club on RNA-Seq data analysis and this is a promising area to work on. Here I want to list some good papers for future reading:
- Julia Salzman, Hui Jiang and Wing Hung Wong (2011), Statistical Modeling of RNA-Seq Data. Statistical Science 2011, Vol. 26, No. 1, 62-83. doi: 10.1214/10-STS343. (We are done with this paper.)
- Turro E, Su S-Y, Goncalves A, Coin LJM, Richardson S and Lewin A (2011). Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biology. 12:R13. journal page. (RNA-seq produces sequence information that can be used for genotyping and phasing of haplotypes, thus permitting inferences to be made about the expression of each of the two parental haplotypes of a transcript in a diploid organism. )
- Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation, Jingyi Jessica Li, Ci-Ren Jiang, James B. Brown, Haiyan Huang, and Peter J. Bickel. ( SLIDE is based on a linear model with a design matrix that models the sampling probability of RNA-Seq reads from different mRNA isoforms. To tackle the model unidentifiability issue, SLIDE uses a modified Lasso procedure for parameter estimation. Compared with deterministic isoform assembly algorithms (e.g., Cufflinks), SLIDE considers the stochastic aspects of RNA-Seq reads in exons from different isoforms and thus has increased power in detecting more novel isoforms. )
- Dalpiaz, D., He, X., and Ma, P. (2012) Bias correction in RNA-Seq short-read counts using penalized regression , Statistics in Biosciences , DOI: 10.1007/s12561-012-9057-6. [Software]
- M. Nicolae and S. Mangul and I.I. Mandoiu and A. Zelikovsky, Estimation of alternative splicing isoform frequencies from RNA-Seq data, Algorithms for Molecular Biology 6:9, 2011, pdf preprint, publisher url, bibtex (In this paper it presents a novel expectation-maximization algorithm for inference of isoform- and
gene-specific expression levels from RNA-Seq data.) - There is a special issue for DNA-Seq, especially the paper: Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data
- Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks
- Sensitive Gene Fusion Detection Using Ambiguously Mapping RNA-Seq Read Pairs (Paired-end whole transcriptome sequencing provides evidence for fusion transcripts. However, due to the repetitiveness of the transcriptome, many reads have multiple high-quality mappings. Previous methods to find gene fusions either ignored these reads or required additional longer single reads. This can obscure up to 30% of fusions and unnecessarily discards much of the data. We present a method for using paired-end reads to find fusion transcripts without requiring unique mappings or additional single read sequencing.) Availability: A C++ and Python implementation of the method demonstrated in this paper is available at http://exon.ucsd.edu/ShortFuse

Recent Comments